
理化学研究所などの研究グループによれば,全ゲノムシークエンスデータから「構造多型(SV/Structural Variation)」を高精度で検出する新しい手法を開発したとのことです。
SVとは,個人間のゲノムの違いのうち50塩基対以上の長さの変異のことで,これを精度よく検出できる単独のツールはこれまで存在しませんでした。
個人ゲノム間に大きな違いをもたらすSVは,発達障害や知的障害を含むさまざまなヒトの疾患・形質の遺伝的要因となることが近年の多くの研究から示されています。また,がんなどの体細胞変異によって引き起こされる疾患においても,SVが関わることを示す多くの研究があります。
今回、共同研究グループは既存の複数のツールを用いてSVを高精度に選別し、この選別過程で抜け落ちたSVを独自の遺伝子型判定手法により回収する、新しいSV検出手法「MOPline」を開発しました。MOPlineを用いて、バイオバンク・ジャパン(BBJ)に登録された約3,300人の全ゲノムシークエンスデータから約134,000(個人当たり約16,000)のSVを検出し、このSVと約18万人のBBJデータを用いて解析をしたところ、多くの疾患や量的形質にSVが関わっていることが明らかになりました。
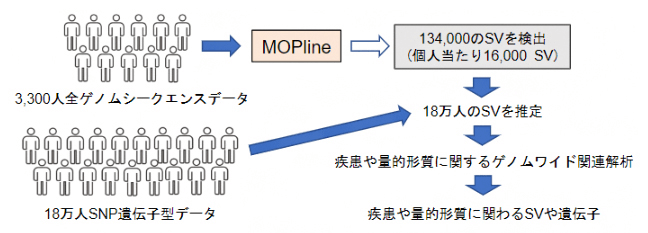
図:構造多型(SV)を用いて疾患や量的形質に関わるSVや遺伝子を究明
本研究成果は、これまで主にSNPを用いて行われていた疾患に関わるゲノム解析を、構造多型を含めた解析に拡張させることを可能にします。
また、MOPlineは、単一および数千の全ゲノムシークエンスデータを用いたSV検出を可能とし、ヒトを含む多様な生物種のSVを検出することができます。このことから、幅広い研究分野において、これまで既存のツールでは不可能であったSVの研究が可能となると期待できます。
今回の研究成果により,これまで発見できなかった疾患や形質の原因となる遺伝子やゲノム変異の同定に貢献することが期待されます。




